
liposarcoma - Liposarcoma
| liposarcoma | |
|---|---|
 | |
| Istopatologia del liposarcoma, colorazione H&E: - | |
| Specialità | Dermatologia , chirurgia generale oncologia |
| Sintomi | Nodulo sottocutaneo, dolore, gonfiore, disfunzione d'organo |
I liposarcomi sono il sottotipo più comune di sarcomi dei tessuti molli , rappresentando almeno il 20% di tutti i sarcomi negli adulti. I sarcomi dei tessuti molli sono neoplasie rare con oltre 150 diversi sottotipi o forme istologiche . I sarcomi, che costituiscono circa l'1% di tutti i tumori maligni dell'adulto, sono tumori maligni che si sviluppano dalle cellule staminali dei tessuti mesenchimali (cioè connettivi) come: gli osteosarcomi che derivano dalle cellule osteoprogenitrici (cioè precursori) le cellule degli osteociti maturi nell'osso tessuti; i fibrosarcomi che originano dalle cellule precursori dei fibrociti nei tessuti connettivi ; ei rabdomiosarcomi che derivano dalle cellule precursori dei miociti nei tessuti muscolari . I liposarcomi derivano dai lipoblasti precursori degli adipociti (cioè le cellule adipose) nei tessuti adiposi (cioè adiposi) . I tessuti adiposi sono distribuiti in tutto il corpo, compresi siti come gli strati profondi e più superficiali dei tessuti sottocutanei , nonché in siti meno accessibili chirurgicamente come il retroperitoneo (cioè lo spazio dietro la cavità addominale ) e il grasso viscerale all'interno della cavità addominale .
Tutti i liposarcomi sono costituiti da almeno alcune cellule che hanno una somiglianza con le cellule adipose quando esaminate per il loro aspetto istopatologico al microscopio. Tuttavia, i liposarcomi hanno diverse forme basate sulle differenze nelle loro presentazioni cliniche (ad es. età, preferenze di genere, sedi di tumori, segni e sintomi ), gravità (cioè potenziale invasione dei tessuti locali, recidiva dopo la rimozione chirurgica e metastasi a livello distale). tessuti), anomalie genetiche , prognosi e regimi di trattamento preferiti. L' Organizzazione Mondiale della Sanità nel 2020 ha riclassificato i liposarcomi in cinque forme più o meno distinte: 1) tumore lipomatoso atipico/liposarcoma ben differenziato; 2) liposarcoma dedifferenziato; 3) liposarcoma mixoide; 4) liposarcoma pleomorfo; e 5) liposarcoma pleomorfo mixoide. ( Pleomorfo indica la presenza di cellule che hanno variazioni anormali e spesso grandi nella loro dimensione e forma e/o nella dimensione e forma dei loro nuclei.)
Mentre le forme di liposarcoma sono classificate come aggressive e maligne o, nel caso del tumore lipomatoso atipico/liposarcoma ben differenziato, come relativamente non aggressive e benigne, tutte e cinque le forme di liposarcoma possono infiltrarsi localmente per danneggiare i tessuti e gli organi vicini, si verificano in siti chirurgicamente inaccessibili adiacenti agli organi vitali (ad es. il retroperitoneo), si ripresentano dopo la rimozione chirurgica e progrediscono verso malattie potenzialmente letali. Gli studi fino ad oggi hanno scoperto che tutte e cinque le forme di liposarcoma, sebbene di solito trattabili almeno inizialmente mediante resezione chirurgica, spesso rispondono solo marginalmente ai regimi di chemioterapia e radioterapia attualmente in uso . I liposarcomi richiedono un'ampia gamma di ulteriori studi per determinare la loro risposta a varie radioterapia , chemioterapia e regimi di trattamento più innovativi usati singolarmente e in varie combinazioni che includerebbero, ove possibile, la rimozione chirurgica.
Forme di liposarcomi
I liposarcomi sono generalmente tumori di grandi dimensioni (>10 cm) ma possono essere di quasi tutte le dimensioni. Si verificano principalmente negli adulti con solo lo 0,7% dei casi che si verificano in quelli di età <16 anni. Negli adulti, i liposarcomi si verificano prevalentemente durante e dopo la mezza età. I casi molto rari che si verificano nei bambini e negli adolescenti vengono diagnosticati prevalentemente come forma di liposarcoma mixoide.
Le cinque forme di liposarcoma devono essere distinte non solo l'una dall'altra, ma anche da alcuni altri tumori dei tessuti molli. Questi altri tumori insieme ad alcune delle loro caratteristiche istopatologiche distintive sono: 1) lipomi displastici (cioè umori benigni che hanno siti di necrosi tissutale e cellule adipose neoplastiche di dimensioni variabili contenenti nuclei di dimensioni/forma variabili ; queste cellule neoplastiche, a differenza della maggior parte delle cellule neoplastiche nei liposarcomi, non sovraesprimono il gene MDM2 ); 2) lipomi fusiformi atipici (cioè tumori benigni con cellule fusiformi lievemente atipiche in uno stroma da fibroso a mixoide misto a lipoblasti vacuolati e adipociti di dimensioni variabili con nuclei atipici; 3) lipomi pleomorfi (cioè tumori benigni caratterizzati da cellule giganti con nuclei sovrapposti); e 4) tumori fibrosi solitari (cioè tumori, fino al 22% dei quali mostrano un comportamento maligno, costituiti da cellule a forma di fuso o ovoidale all'interno di uno stroma di fondo collagene mescolato con vasi sanguigni con una caratteristica forma a corna di cervo).
Tumore lipomatoso atipico/liposarcoma ben differenziato
Insieme, i tumori lipomatosi atipici (ALT) e i liposarcomi ben differenziati (WDL) rappresentano dal 40% al 45% di tutti i liposarcomi. Raramente, se non mai, metastatizzano e quindi sono considerati tumori benigni o premaligni . Tuttavia, sono localmente invasivi e possono trasformarsi in un liposarcoma più aggressivo e potenzialmente metastatizzante, cioè un liposarcoma dedifferenziato. Inoltre, un tumore lipomatoso atipico/liposarcoma ben differenziato rimosso chirurgicamente può ripresentarsi come liposarcoma dedifferenziato.
Presentazione

ALT e WDL sono considerati tumori virtualmente identici, tranne per il fatto che per definizione gli ALT designano tumori che si sviluppano nelle braccia o nelle gambe mentre i WDL designano tumori che si sviluppano in siti meno accessibili chirurgicamente come i tessuti molli profondi e centrali del retroperitoneo , regione paratesticolare ( cioè area all'interno del scroto compresi i testicoli , funicolo spermatico , tunica testicoli , epididimo , e appendice di testicolo ), cavità orale e delle orbite . Questa terminologia ha implicazioni prognostiche: meno del 7% dei tumori ALT si converte in liposarcomi dedifferenziati entro un tempo mediano di 7 anni mentre il 17% dei tumori WDL si converte in questo liposarcoma più maligno entro un tempo mediano di 8 anni. I tumori ALT e WDL (di seguito denominati ALT/WDL) si presentano tipicamente negli individui di mezza età e più anziani come masse che si ingrandiscono lentamente che tendono ad essere più grandi e in uno stadio più avanzato quando si trovano nei tessuti profondi. Questi tumori di solito non sono dolorosi e se localizzati superficialmente, facilmente evidenti; possono anche causare edema esteso (ossia gonfiore dovuto all'accumulo locale di liquidi) nelle zone interessate come la coscia (vedi figura a lato) per la loro invasione nei vasi sanguigni e/o linfatici che drenano la sede del tumore. I tumori ALT/WDL profondi possono essere asintomatici ma, a seconda della loro localizzazione, producono gravi segni e/o sintomi di disfunzione in uno qualsiasi dei vari organi in cui si infiltrano. Questi organi includono quelli vicini o nel retroperitoneo (es. intestino, rene e ureteri renali ); la regione paratesticolare; il mediastino (es. trachea e bronchi maggiori del polmone ); e la testa (es. lo spazio retrobulbare dietro il globo oculare).
Patologia

Dal punto di vista istopatologico, i tumori ALT/WDL sono suddivisi in varianti adipocitiche/lipoma-simili, sclerosanti e infiammatorie, con le più comuni adipociti/lipoma-simili. I tumori ALT/WDL adipocitici/lipoma-simili sono costituiti da lobuli di cellule adipose mature variamente intersecate con setti fibrosi irregolari (vedere la microfotografia colorata con H&E adiacente ). I tumori sclerosanti ALT/WDL, la seconda variante più comune, si sviluppano principalmente nelle aree retroperitoneali e paratesticolari; è costituito da cellule stromali sparse e atipiche all'interno di uno sfondo di tessuto stromale collagene (cioè contenente collagene ) . Rari lipoblasti contenenti vacuolo popolano questo tessuto. I tumori infiammatori ALT/WDL sono la variante più rara. si verificano più frequentemente nel retroperitoneo e sono costituiti da cellule infiammatorie croniche , ad esempio linfociti e plasmacellule più occasionali follicoli simili a linfonodi disseminati in un tessuto di fondo contenente cellule adipose .
Genetica
Le cellule neoplastiche nei tumori ALT/WDL contengono uno o più cromosomi marcatori soprannumerari piccoli extra a forma di anello (sSMC) o un cromosoma marcatore gigante anormale (cioè un cromosoma precedentemente normale reso anormale dalla duplicazione di parti del proprio o uno o più materiale genetico di altri cromosomi). Questi cromosomi anormali contengono copie extra del braccio lungo del cromosoma 12 (chiamato anche braccio q ) nelle bande da 13 a 15. Questo tratto del cromosoma 12 include il proto-oncogene MDM2 (un gene potenzialmente cancerogeno quando sovraespresso ) situato nella banda 15 e CDK4 (un gene che quando sovraespresso favorisce lo sviluppo di vari tumori) localizzato alla banda 14.1. L' amplificazione (cioè l'aumento delle copie di un gene senza un aumento proporzionale di altri geni) di questi due geni è un indicatore altamente sensibile e specifico che un liposarcoma è un ALT/WDL o un liposarcoma dedifferenziato piuttosto che qualsiasi altra forma di liposarcoma o lipoma . Oltre ai geni MDM2 e CDK4 , questa area cromosomica della banda 13-15 contiene anche i geni TSPAN31 e HMGA2 che, quando sovraespressi, sono associati a vari tumori e/o tumori. È stato suggerito che uno o più di questi geni sovraespressi promuovano e/o contribuiscano allo sviluppo e/o alla progressione dei tumori ALT/WDL.
Diagnosi
La diagnosi dei tumori ALT/WDL viene effettuata in base alle caratteristiche delle loro presentazioni cliniche, istopatologia e reperti genetici. In particolare, il rilevamento nelle cellule tumorali ALT/WDL di un gene MDM2 o CDK4 sovraespresso o la presenza della specifica sSMC associata a ALT/WDL o del cromosoma marcatore gigante (come definito dal sequenziamento del DNA di nuova generazione , dall'ibridazione genomica comparativa e/ o analisi di bande G citogenetiche altamente specializzate ) supporta fortemente la diagnosi di ALT/WDL o liposarcoma dedifferenziato. La presentazione clinica e le differenze istopatologiche tra le ultime due forme di liposarcoma di solito aiutano a distinguerle.
Trattamento e prognosi
I tumori ALT/WDL sono trattati mediante resezione chirurgica radicale per rimuovere tutti i tessuti neoplastici tumorali. Tuttavia, questi tumori si ripresentano localmente nel 30-50% dei casi. Le recidive si verificano più spesso nei tumori localizzati in siti meno accessibili come quelli del retroperitoneo, del mediastino e del funicolo spermatico. Questi tumori meno valutabili chirurgicamente tendono a ripresentarsi ripetutamente e alla fine possono causare la morte a causa dei loro effetti dannosi sugli organi vitali. Mentre i tumori ALT/WDL hanno pochissimo potenziale di metastatizzare , circa il 10% si convertirà in una forma di liposarcoma apertamente maligna e potenzialmente metastatizzante, il liposarcoma dedifferenziato. Il tempo mediano per questa trasformazione maligna è di circa 7-9 anni. Inoltre, un ALT/WDL rimosso chirurgicamente può ripresentarsi dopo un intervallo variabile come liposarcoma dedifferenziato. Un ampio studio randomizzato controllato che confrontava la radioterapia seguita dalla chirurgia alla sola chirurgia nei tumori ALT/WDL ha riscontrato poche differenze tra i due regimi. Studi più piccoli che impiegano inibitori selettivi dei prodotti proteici dei geni CDK4 o MDM2 implicati in ALT/WDL hanno mostrato nel migliore dei casi solo effetti modesti. Sono in corso ulteriori studi che utilizzano questi o regimi di trattamento completamente nuovi. Uno studio di revisione nel 2012 ha riportato che i tassi di sopravvivenza a 5 e 10 anni degli individui con ALT/WDL sono rispettivamente del 100% e dell'87%.
Nuove terapie
Le nuove terapie di ALT/WDL sono le stesse elencate nella sezione Nuove terapie di Liposarcoma dedifferenziato.
Liposarcoma dedifferenziato
I liposarcomi dedifferenziati sono tumori maligni che in circa il 10% dei casi si sviluppano in un tumore lipomatoso atipico esistente/liposarcoma ben differenziato (ALT/WDL) o nel sito in cui un tumore ALT/WPL è stato rimosso chirurgicamente. Gli individui con una diagnosi de novo di questo tumore possono aver avuto un ALT/WDL che è progredito in un liposarcoma dedifferenziato ma non è stato rilevato perché si è sviluppato in modo asintomatico in un sito altamente sequestrato come il retroperitoneo o la cavità addominale. Molte delle caratteristiche cliniche e genetiche dei tumori del liposarcoma dedifferenziato sono simili a quelle riscontrate nei tumori ALT/WDL.
Presentazione
I lipoosarcomi dedifferenziati (DDL) si verificano più frequentemente negli adulti di mezza età e negli anziani con un picco di incidenza tra la sesta e l'ottava decade. Raramente, questi tumori si sono sviluppati nei bambini e negli adolescenti. I tumori DDL si verificano più comunemente nello spazio retroperitoneale ma, simili a ALT/WDL, possono verificarsi nelle estremità, nell'area paratesticolare, nel mediastino, nella testa o nel collo. Meno dell'1% di tutte le DDL si sviluppa nei tessuti molli superficiali o nella cavità oculare. Alla presentazione, i tumori DDL sono tipicamente indolori, di grandi dimensioni, possono essersi ingranditi lentamente e progressivamente per anni e nelle radiografie di routine contengono aree di deposizione di calcio (esemplificate dalla Fig. 1 nella sezione Istopatologia dei liposarcomi). Meno comunemente, gli individui affetti presentano segni e/o sintomi dovuti all'impatto del tumore su un organo (ad es. dolore addominale causato dal blocco dell'intestino o dall'ostruzione del tratto urinario causata dal blocco dell'uretra ). Molto raramente, gli individui con DDL presentano uno o più segni o sintomi di infiammazione cronica (vedi sintomi B ) e/o una delle sindromi endocrine , neurologiche , mucocutanee , ematologiche o paraneoplastiche tissutali . I segni ei sintomi dell'infiammazione cronica e delle varie sindromi paraneoplastiche sono causati dalla secrezione da parte dei tumori di citochine , ormoni , prostaglandine e/o altri agenti ad azione sistemica; scompaiono completamente dopo che il DDL è stato trattato con successo.
Patologia
L' aspetto istopatologico dei tumori DDL (vedi Fig. 2 nella sezione Istopatologia dei liposarcomi sotto) varia ampiamente ma mostra più frequentemente le caratteristiche dei sarcomi pleomorfi indifferenziati (che sono tumori densamente popolati con cellule di dimensioni e forma variabili contenenti nuclei di dimensioni e forma variabili ) o sarcomi a cellule fusiformi (che sono tumori costituiti da cellule a forma di fuso in uno sfondo di tessuto connettivo ). Diverse parti dei tumori DDL mostrano spesso variazioni nell'aspetto dei loro tessuti connettivi di fondo: questi tessuti possono essere mixoidi (cioè costituiti da una sostanza chiara simile al muco che, quando colorata con un metodo standard di colorazione H&E , appare più blu o viola rispetto al rosso colore dei tessuti normali) o myxocollagenous (ovvero alta collagene contenuto di fibre in uno sfondo mixoide), e, in ~ 5% dei casi, hanno aree di osteoide (vedi Fig. 1 nel sotto istopatologia sezione liposarcomi) o cartilagineo materiale . Questi tumori mostrano anche grandi variazioni nel loro contenuto cellulare. Ad esempio, fino al 10% dei tumori DDL ha aree con istopatologia ALT/WDL e rari casi di DDL hanno aree contenenti spirali di cellule piatte simili a meningoteli.
Genetica
Le cellule neoplastiche sia in DDL che in ALT/WDL portano cromosomi marcatori soprannumerari piccoli simili (sSMC) e/o cromosomi marcatori giganti che contengono parti extra del braccio q del cromosoma 12 nelle bande da 13 a 15. Questa area cromosomica include due geni associati allo sviluppo del tumore , i geni MDM2 e CDK4 . La presenza di copie extra di questi due geni e/o dei loro prodotti proteici in eccesso è un indicatore altamente sensibile e specifico che un tumore lipomatoso è un ALT/WDL o DDL piuttosto che qualche altro tipo di tumore lipomatoso. Si sospetta che la sovraespressione dei geni MDM2 e CDK e/o altro materiale genetico nelle sSMC o nei cromosomi marcatori giganti promuova lo sviluppo e/o la progressione dei tumori DDL e ALT/WDL. Altri geni nella sMMC e nel cromosoma marcatore gigante che sono anche sovraespressi nelle cellule neoplastiche ALT/WDL e DDL includono HMGA2 , CPM , YEATS4 e DDIT3 . Rispetto alle cellule neoplastiche ALT/WDL, tuttavia, le cellule neoplastiche DDL: 1) esprimono livelli più elevati dei geni nei due cromosomi anormali; questo può contribuire alla progressione di ALT/WDL in DDL; e 2) livelli più elevati di prodotti genici sul braccio lungo del cromosoma 1 alla banda 32, il braccio lungo del cromosoma 6 alla banda 33 e, in ~25% dei casi, il braccio corto del cromosoma 1 alla banda 32,2 che contiene il Gene JUN (questo gene è sovraespresso in DDL ma non in ALT/WDL). Poiché il prodotto del gene JUN , c-jun , inibisce la morte cellulare e promuove la proliferazione cellulare, la sua sovrapproduzione può contribuire alla progressione di ALT/WDL a DDL e/o alla malignità delle cellule neoplastiche DDL. Il profilo di espressione genica (cioè la misurazione dell'espressione dei prodotti di migliaia di geni prodotti da cellule, tessuti o tumori) ha rivelato che la differenziazione delle cellule degli adipociti e le vie metaboliche in ALT/WDL sono sovraregolate mentre la proliferazione cellulare e le vie di risposta al danno del DNA sono sovraregolate in DDL.
Diagnosi
L'istopatologia del DDL è spesso insufficientemente chiara per fare una diagnosi definitiva. Tuttavia, la diagnosi di DDL è supportata in individui: i cui tumori contengono ALT/WDL mescolati con componenti istologiche DDL; con storie di precedenti ALT/WDL; o che presentano un liposarcoma retroperitoneale (il DDL costituisce circa il 57% di tutti i liposarcomi retroperitoneali). I tumori DDL solo raramente (<1% dei casi) si presentano come tumori cutanei superficiali; hanno quasi 5 volte meno probabilità di ALT/WDL di verificarsi nell'orbita dell'occhio; e sono estremamente rari nei bambini. Il rilevamento dell'amplificazione MDM2 delle cellule tumorali è il gold standard diagnostico per distinguere il WDL da lipomi, lipomi displastici, sarcomi a cellule fusate atipiche, lipomi pleomorfi e tumori fibrosi solitari. In alternativa, il rilevamento nelle cellule tumorali di un gene CDK4 sovraespresso o la presenza delle specifiche sSMC associate a ALT/WDL o del cromosoma marcatore gigante supportano fortemente la diagnosi di DDL o ALT/WDL. La presentazione clinica, l'istopatologia e le differenze genetiche (ad es. la sovraespressione delle cellule tumorali del gene cJUN favorisce fortemente la diagnosi di DDL rispetto ad ATL/WDL) tra queste ultime due forme di liposarcoma di solito aiutano a distinguerle.
Trattamento e prognosi
La resezione chirurgica completa è solitamente il trattamento di prima linea raccomandato per i tumori DDL localizzati. Tuttavia, studi emergenti suggeriscono che i pazienti con tumori DDL che sono limitati a un'estremità o al tronco e hanno una sopravvivenza globale correlata al tumore prevista a 10 anni pari o inferiore al 51% hanno esiti migliori quando si aggiunge la chemioterapia (ad es. doxorubicina più ifosfamide ). loro regimi chirurgici. Per queste forme localizzate di DDL, può essere presa in considerazione anche la radioterapia perioperatoria secondo le linee guida del National Comprehensive Cancer Network .
La DDL retroperitoneale è la forma più comune, chirurgicamente non valutabile e grave di DDL: ha un tasso di recidiva del 66% e un tasso di sopravvivenza globale a cinque anni del 54%. L'opzione di trattamento principale per la DDL retroperitoneale è la resezione chirurgica. Uno studio clinico di fase III ha riscontrato poche differenze nei risultati della radioterapia seguita da resezione chirurgica rispetto alla sola resezione chirurgica nel trattamento del DDL retroperitoneale. In altri studi clinici di fase III, pazienti con DDL con tumori retroperitoneali e/o metastatici inaccessibili sono stati trattati con chemioterapia di prima linea confrontando doxorubicina a doxorubicina più ifosfamide o doxorubicina a gemcitabina più docetaxel . Anche altri studi hanno esaminato il valore di vari regimi chemioterapici. Questi studi hanno spesso trovato poche differenze nei tempi di sopravvivenza globale nei loro confronti, ma hanno mostrato alcuni miglioramenti nella sopravvivenza libera da progressione e in altri parametri clinici. Sulla base di questi studi, una terapia di prima linea raccomandata per i tumori retroperitoneali e altri tumori DDL non valutabili chirurgicamente o metastatici è il trattamento con un regime chemioterapico a base di antracicline o, nei casi resistenti al tumore o recidivanti, la chemioterapia con eribulina . Una revisione condotta nel 2020 ha riportato che i tempi di sopravvivenza mediani per DDL di basso grado istopatologico e di alto grado istopatologico sono rispettivamente di 113 mesi e 48 mesi. Sono necessari ulteriori studi per fornire prove sull'efficacia della radioterapia, della chemioterapia e delle nuove terapie in tutte le varietà di DDL.
Nuove terapie
Diversi nuovi regimi terapeutici per il DDL ei casi più aggressivi o comunque problematici di ALT/WDL sono attualmente in fase di sperimentazione clinica . È in corso uno studio clinico di fase II che indaga su abemaciclib in pazienti con DDL pretrattato o non trattato. L'analisi preliminare ha mostrato che questo inibitore del prodotto dei geni CDK4 e CDK6, gli enzimi della proteina chinasi specifici della serina/treonina hanno prodotto un tempo di sopravvivenza mediano senza progressione prolungato di 30,4 settimane. Uno studio clinico di fase III multicentrico, randomizzato, in doppio cieco, controllato con placebo su abemaciclib è nella sua fase attiva e presto (come dichiarato a luglio 2021) inizierà a reclutare 108 individui con DDL avanzato, ricorrente e/o metastatico. Lo studio è sponsorizzato dalla Sarcoma Alliance for Research through Collaboration in collaborazione con Eli Lilly and Company . Ribociclib , anche un inibitore dei geni CDK4 e CDK6 , in combinazione con un inibitore di mTOR , everolimus è in uno studio clinico di fase II in individui con DDL avanzato o leiomiosarcoma . Uno studio di registrazione di fase III (ovvero un ampio studio di conferma inteso a stabilire un profilo beneficio/sicurezza accettabile al fine di ottenere l'approvazione normativa per un'indicazione precisamente definita) sta valutando la sicurezza e l'efficacia del milademetan rispetto alla trabectedina in pazienti con malattia non resecabile (es. si ritiene che la resezione causi morbilità o mortalità inaccettabili) o DDL metastatico che è progredito con 1 o più precedenti terapie sistemiche, inclusa almeno 1 terapia a base di antracicline. Lo sponsor, Rain Therapeutics Inc, sta attualmente reclutando 160 persone per lo studio. Un'altra fase III di sperimentazione clinica sta valutando l' MDM2 inibitore milademetan contro trabectedina , un bloccante del oncogenica fattore di trascrizione FUS-CHOP , in MDM2 -overexpressing ALT / WDL e DDL. Milademetan ha mostrato una tossicità gestibile e una certa attività risultante in una malattia stabile e/o alcune risposte parziali nel DDL.
Liposarcoma mixoide
Presentazione
Il liposarcoma mixoide (MLS), che include un tipo di liposarcoma chiamato liposarcoma a cellule rotonde, rappresenta circa il 30% di tutti i liposarcomi. Ha un picco di incidenza nella quarta e quinta decade degli individui con una predominanza maschile nella maggior parte degli studi. Sebbene non comune nei bambini e negli adolescenti, la MLS è la forma di liposarcoma più comune diagnosticata in questi gruppi di età. La MLS si presenta tipicamente come una massa grande (da 1 a 39 cm; media 12 cm), mobile, ben circoscritta e indolore che si è sviluppata da 1 settimana a 15 anni prima della diagnosi. I tumori MLS sono localizzati nei tessuti molli profondi delle cosce (65-80% dei casi), parte inferiore delle gambe (10-15% dei casi), retroperitoneo (8% dei casi) e braccia (5% dei casi). In circa un terzo dei casi, questi tumori metastatizzano in altri siti dei tessuti molli (ad es. retroperitoneo, torace o altre estremità), osso scheletrico e/o polmone. Gli individui possono presentare queste metastasi, in particolare quelle ossee; è stato raccomandato che i pazienti debbano essere testati alla presentazione per metastasi ossee mediante imaging medico , inclusi raggi X , scansioni TC e/o risonanza magnetica .
Patologia
Le analisi istopatologiche della MLS (vedi Figg. 3 e 4 nella sezione Istopatologia dei liposarcomi sotto) rivela cellule sparse in una matrice mixoide (cioè uno sfondo di tessuto connettivo che appare più blu o viola rispetto al colore rosso del normale tessuto connettivo quando questi tessuti sono adeguatamente preparati, colorati con H&E e osservati al microscopio). Queste cellule sono lipoblasti , alcuni dei quali sono a forma di anello con sigillo (una forma che suggerisce che la cellula possa essere neoplastica), di forma ovale o di forma rotonda. I tumori MLS possono essere ipercellulari e contenere fogli solidi di cellule rotonde che comprendono almeno il 5% di tutte le cellule o una bassa cellularità popolata da cellule che hanno nuclei blandi e cellule rotonde <5% in uno sfondo di capillari ricurvi che ricordano un reticolo di pollo. I tumori che contengono almeno il 5% di cellule rotonde sono classificati come di alto grado mentre quelli con <5% di cellule rotonde sono classificati come di basso grado. I tumori MLS di alto grado in genere hanno un decorso clinico più aggressivo rispetto ai tumori MLS di basso grado.
Genetica
Le cellule tumorali MLS sono virtualmente definite dalla loro espressione di un gene di fusione FUS-DDIT3 (chiamato anche gene chimerico ) che si verifica in >95% dei casi o di un gene di fusione EWSR1-DDIT3 che si verifica nel restante <5% dei casi. Il gene di fusione FUS-DDIT3 si forma come risultato di una traslocazione (denominata t(12:16)(q13:p11)) tra il sito del gene DDIT3 sulla banda 12 del braccio q del cromosoma 12 e il sito del gene FUS a banda 11 sul braccio corto del cromosoma 16 (chiamato anche braccio p ). Il prodotto della proteina di fusione (chiamata anche proteina chimerica) di questo gene oncogene chimerico , FUS-DDIT3, è noto per arrestare la maturazione delle cellule adipose e promuovere la neoplasia. Il gene di fusione EWSR1-DDIT3 (denominato t(12;22)(q13;q12)) deriva da una traslocazione del gene EWSR1 situato nella banda 12.2 sul braccio q del cromosoma 22 con il gene DDIT2 . Il prodotto della proteina di fusione del gene EWSR1-DDIT3 , come la proteina di fusione FUS-DDIT3, promuove la neoplasia. Nonostante queste relazioni geniche di fusione, sono necessari ulteriori studi per definire il loro contributo allo sviluppo e/o al mantenimento dei tumori MLS.
Diagnosi
I tumori MLS di basso grado e di grado intermedio possono essere identificati istologicamente dalla loro morfologia classica di caratteristica vascolarizzazione a filo di pollo disseminata in uno stroma mixoide. Tuttavia, i tumori MLS di alto grado possono essere difficili da distinguere da altre neoplasie a cellule rotonde, in particolare i tumori MLS di alto grado che consistono in una morfologia cellulare diffusa e/o pura a cellule rotonde in misura tale da oscurare questo classico pattern vascolare-mixoide. Il rilevamento di un riarrangiamento del gene DDIT3 con il gene FUS o EWSR1 mediante ibridazione in situ o immunoistochimica o le trascrizioni di fusione dell'RNA di questi geni mediante reazioni a catena della polimerasi in tempo reale conferma la diagnosi di casi di alto grado e ambigui di basso grado o tumori MLS di grado intermedio.
Trattamento e prognosi
La MLS è stata tipicamente trattata mediante resezione chirurgica, ma può richiedere interventi più radicali, ad esempio l'amputazione dell'arto può essere necessaria quando il fascio neurovascolare di un arto è compromesso. È stato riportato che il rischio post-chirurgico di recidiva entro 3 anni dall'intervento chirurgico è di ~ 15% quando non tutto il tumore viene rimosso e ~ 10% quando la rimozione del tumore è completa. L'aggiunta della radioterapia alla resezione chirurgica ha migliorato il controllo locale dei tumori MLS ed è stata raccomandata per il trattamento della MLS non resecabile e ricorrente. Tuttavia, sono necessari ulteriori studi per determinare il valore della radioterapia nel trattamento delle varie varietà di MLS. I regimi chemioterapici che utilizzano ifosfamide , un'antraciclina come daunorubicina , dacarbazina e/o trabectedina sono stati trovati utili: uno studio clinico di fase III ha mostrato tempi di sopravvivenza libera da progressione nei pazienti con MLS trattati con trabectedina o dacarbazina rispettivamente di 5,6 e 1,5 mesi. Nel 2015 la Food and Drug Administration ha approvato la trabectedina per l'uso nei liposarcomi non resecabili e metastatici.
Complessivamente, il tasso di sopravvivenza a 10 anni degli individui con MLS è stato del 77%, un tasso di sopravvivenza notevolmente più lungo rispetto ad altre forme di liposarcoma. Rispetto alla MLS a basso rischio, la MLS ad alto rischio (rischio definito dal contenuto di cellule rotonde tumorali e/o altri indicatori prognostici sfavorevoli) è associata a un aumento dei tassi di metastasi e quindi a un tempo di sopravvivenza più breve. L'aumento delle dimensioni del tumore (≥ 10 cm) è fortemente associato a una MLS di grado più elevato e quindi a un tempo di sopravvivenza più breve. Altri fattori che sono stati associati a esiti sfavorevoli nella MLS includono la presenza di necrosi tumorale, età > 45 anni, sovraespressione del gene P53 e sesso maschile. Anche la forma a cellule rotonde dei liposarcomi mixoidi sembra avere una prognosi relativamente sfavorevole: in varie revisioni retrospettive, il liposarcoma mixoide è stato generalmente trovato di basso grado e quindi relativamente sensibile alla chemioterapia, mentre il labbarcoma mixoide di alto grado (cioè a cellule rotonde) aveva tassi più elevati di metastasi, si è comportato in modo più aggressivo e non ha risposto bene alla chemioterapia.È importante notare, tuttavia, che quasi tutti i casi di liposarcomi mixoidi nei pazienti pediatrici hanno avuto una prognosi eccellente.
Nuove terapie
Un agonista PPAR-γ (cioè attivatore), efatutazone, è stato studiato in un piccolo studio di fase I su individui con varie neoplasie in stadio avanzato. Il farmaco ha prodotto una risposta marcatamente durevole in una persona con MLS, suggerendo che gli agonisti PPAR-γ sarebbero utili per il trattamento di questa malattia. Uno studio clinico di stadio II condotto in Italia sta esaminando gli effetti di una trabectedina più pioglitazone (un altro agonista PPAR-γ) in individui con tumori MLS stabili. Lo studio prevede due fasi sequenziali. La prima fase esamina la risposta dei pazienti trattati per un minimo di 4 cicli con la sola trabectedina. Se si ottiene una malattia stabile, la seconda fase esaminerà gli effetti di un ulteriore trattamento dei pazienti che inizialmente rispondono con una combinazione di trabectedina e pioglitazone. È in fase di completamento uno studio clinico di stadio II per valutare l'efficacia di sirolimus (un inibitore di MTOR ; sirolimus è anche noto come rapamicina) più ciclofosfamide (un farmaco chemioterapico) nella MLS metastatica o non resecabile. Uno studio clinico di fase II sta reclutando pazienti per valutare sintilimab (un anticorpo monoclonale IgG4 umano diretto contro la proteina 1 della morte cellulare programmata situata sulla superficie delle cellule) in combinazione con due farmaci chemioterapici, doxorubicina e ifosfamide , come trattamento di prima linea della sarcomi tissutali compreso MLS.
Le cellule T sono state ingegnerizzate geneticamente per colpire l' antigene MAGE-A4 espresso su un peptide contenente HLA-A*02 MAGE-A4 situato sulla superficie delle cellule neoplastiche in alcuni tipi di tumori. Queste cellule ingegnerizzate (chiamate cellule ADP-A2M4-T) hanno attaccato e ucciso varie cellule tumorali umane in coltura recanti questo antigene e, in uno studio clinico di fase 1, hanno ridotto vari tipi di tumori solidi in pazienti i cui tumori contenevano cellule neoplastiche che esprimevano questo antigene. Uno studio clinico di fase II sta ora reclutando individui per studiare l'efficacia e la sicurezza delle cellule T ADP-A2M4 (progettate dalle cellule T del ricevente) in pazienti HLA-A*02-postitivi con MSGE-4 metastatico o inoperabile in stadio avanzato - tumori MLS positivi.
Liposarcoma pleomorfo
Presentazione
I liposarcomi pleomorfi (PLS), che rappresentano dal 5% al 10% di tutti i casi di liposarcoma, sono tumori degli adipociti a crescita rapida, generalmente di grandi dimensioni (> 5 cm) e indolori ma altamente maligni. Si verificano principalmente in individui >50 anni con una predominanza nelle femmine. I tumori PLS si trovano raramente nei bambini. Tumori PLS presenti in una gamba o un braccio (65% dei casi), retroperitoneo o addome (15% dei casi), o in rari casi parete del tronco, funicolo spermatico , aree della testa e del collo, parete toracica, cavità pelvica , pleure polmonari , pericardio e colonna vertebrale. Questi tumori sono solitamente localizzati nei tessuti molli profondi con solo il 25% dei casi che si presentano nei tessuti sottocutanei. Rari casi di PLS si sono presentati in individui con le sindromi di Li-Fraumeni o Muir-Torre , due malattie genetiche ereditarie che predispongono le persone affette a sviluppare vari tipi di cancro.
Patologia
L'istopatologia dei tumori PLS spesso consiste in aree che ricordano il liposarcoma mixoide mescolate con aree contenenti cellule indifferenziate. Questi tumori sono marcati ipercellulari e contengono almeno alcuni lipoblasti di forma variabile che hanno nuclei pleomorfi. Le aree di necrosi sono comuni, le cellule giganti, alcune delle quali sono multinucleate e/o contengono neutrofili inglobati , sono occasionalmente presenti e le goccioline ialine possono essere viste in alcune cellule così come sparse a livello extracellulare in tutto il tumore. La componente indifferenziata di questi tumori è più spesso costituita da cellule fusiformi, con il 25% dei casi che mostra cellule con morfologia cellulare epitelioide . Questi tumori hanno almeno alcuni focolai con un'istopatologia simile agli istiocitomi di tipo mixofibrosarcoma di alto grado , un tumore precedentemente chiamato istiocitoma fibroso mixoide maligno.
Genetica
Le cellule neoplastiche della PLS contengono varie anomalie genetiche e cromosomiche: il gene TP53 è deleto o mutato nel 17-60% dei casi; il gene RB1 è deleto nel 60% dei casi; e il gene Neurofibromin 1 viene perso inattivando mutazioni nell'8% dei casi o in casi più rari per delezione attorno alla sua posizione nella banda 11.2 sul braccio lungo del cromosoma 12. Queste cellule possono anche mostrare guadagni nel materiale genetico intorno: bande 12 -15 sul braccio corto del cromosoma 5; banda 21 sul braccio corto del cromosoma 1; e la banda 22 sul braccio lungo del cromosoma 7. Le alterazioni nel numero di copie geniche indotte da queste anomalie sono simili a quelle osservate nel tipo myxofibrosarcoma degli istiocitomi . Il ruolo (i) di questi cambiamenti nei numeri di copie del gene nella promozione della PLS non è stato definito. Pertanto, il PLS è diverso da altri liposarcomi in quanto le sue cellule neoplastiche hanno un genoma complesso senza alterazioni genomiche caratteristiche o geni identificabili che guidano la malattia. La rilevazione di alterazioni nell'espressione dei geni TP53, RB1 e neurofibromina 1 , nonché di altri geni meno comunemente alterati nella PLS (ad es. PIK3CA , tirosina-proteina chinasi SYK , PTK2B , EPHA5 e ERBB4 ), può aiutare a supportare ma non definire chiaramente un tumore come PLS. L'estensione delle estremità dei telomeri cromosomiche mediante meccanismi patologici denominati allungamento alternativo dei telomeri si verifica nelle cellule neoplastiche di circa l'80% dei casi di PLS, ma è molto meno comune o non si osserva nelle altre quattro forme di liposarcoma.
Diagnosi
La diagnosi di PLS dipende dalla sua presentazione, istopatologia e genetica. L'istopatologia della PLS spesso assomiglia molto a quella del mixofibrosarcoma, ma si distingue da quel tumore per il suo contenuto di lipoblasti pleomorfi.
Trattamento e prognosi
La resezione chirurgica radicale è il trattamento principale per la PLS; è anche un importante intervento palliativo per alleviare i sintomi dovuti alla compressione di organi e tessuti. La chirurgia può richiedere la rimozione di un intero organo compresso come il rene o il colon. Indipendentemente da questo intervento chirurgico, tuttavia, i tassi di recidiva locale sono molto alti. Gli usi della chemioterapia e/o della radioterapia in combinazione con la chirurgia radicale non hanno dimostrato di prolungare la sopravvivenza e sono considerati interventi controversi. Il National Comprehensive Cancer Network raccomanda il trattamento per gli individui con PLS localizzato ad alto rischio mediante resezione chirurgica completa, quando possibile, in combinazione con la radioterapia. Gli individui con malattia metastatica sono stati trattati con chemioterapia (ad es. doxorubicina più ifosfamide o eribulina ) simile ai regimi utilizzati per il liposarcoma dedifferenziato (vedere la sezione precedente sul trattamento di questo tipo di liposarcoma) Circa il 20% dei tumori PLS metastatizza in siti distanti, il più comuni dei quali sono polmone (82% delle metastasi), fegato (18% delle metastasi) e ossa o pancreas (18% delle metastasi). I tassi di sopravvivenza della PLS a 1, 3 e 5 anni sono riportati rispettivamente del 93%, 75% e 29%. I tumori localizzati nella posizione centrale del tronco, più grandi di 10 cm, profondamente radicati o contenenti aree di necrosi hanno prognosi peggiori.
Liposarcoma pleomorfo mixoide
Il liposarcoma pleomorfo mixoide (originariamente chiamato liposarcoma mixoide pleomorfo) è stato descritto per la prima volta in un ampio studio del 2009 sui liposarcomi. Sebbene inizialmente considerato come una possibile variante dei liposarcomi mixoidi con caratteristiche pleomorfe , l' Organizzazione Mondiale della Sanità (2020) lo ha classificato come una nuova e distinta forma dei liposarcomi. Questa classificazione si basava sui risultati che i liposarcomi pleomorfi mixoidi, pur avendo caratteristiche istopatologiche simili ai liposarcomi mixoidi, avevano caratteristiche genetiche e molecolari cliniche e, soprattutto, critiche che differivano dal mixoide e dalle altre tre forme di liposarcoma.
Presentazione
Il liposarcoma pleomorfo mixoide (MPL) è una forma eccezionalmente rara e altamente aggressiva dei liposarcomi che si sviluppa in bambini, adolescenti, giovani adulti e, in uno studio più recente, individui di età superiore ai 50 anni. I tumori MPL si presentano come masse profonde dei tessuti molli che spesso si trovano nel mediastino e, meno spesso, nelle estremità, nella testa e nel collo, nella cavità addominale o nel tronco. Almeno due casi di MPL si sono presentati in individui con la sindrome di Li-Fraumeni , una malattia genetica ereditaria che predispone gli individui a sviluppare vari tipi di cancro.
Patologia
All'analisi istopatologica, i tumori MPL sono costituiti da aree che assomigliano al liposarcoma mixoide convenzionale; queste aree, che rappresentano il 30-50% delle aree tumorali totali, hanno un'abbondante matrice mixoide, una vascolarizzazione capillare ben sviluppata, cellule blande che sono rotonde e/o leggermente fusiformi, lipoblasti vacuolati e cellule multinucleate a forma di piccoli fiori. Tuttavia, queste aree contengono anche una dispersione di cellule altamente pleomorfe che mostrano maggiori gradi di allargamento nucleare e irregolarità rispetto alle cellule osservate nei tumori del liposarcoma mixoide. Altre aree dei tumori MPL sono più cellulari e sono costituite da lipoblasti a rapida crescita e altamente pleomorfi .
Genetica
Le cellule neoplastiche in MPL non esprimono i geni di fusione FUS-DDIT3 o EWSR1-DDIT3 che sono espressi dalle cellule neoplastiche in >95% o <5%, rispettivamente, dei casi di fibrosarcoma mixoide. L'inattivazione del gene oncosoppressore RB1 a causa della sua delezione o soppressione patologica si riscontra in tutti i casi MPL. Le cellule neoplastiche MPL hanno comunemente anche altre alterazioni nei loro cromosomi. Possono mostrare guadagni anomali in parte del materiale genetico normalmente presente sui cromosomi 1, 6, 7, 8, 19, 21 e/o X e perdite nel materiale genetico normalmente presente sui cromosomi 2, 3, 4, 5, 10 , 11, 12, 13, 14, 15, 16, 17 e / o 22. il materiale genetico perso in banda 14 sul braccio lungo del cromosoma 13 include non solo il RP1 gene ma anche la RCBTB2 , DLEU1 , e ITM2B geni. A causa della sua rarità e della definizione più recente, le caratteristiche molecolari e l'importanza di queste anomalie genetiche devono ancora essere completamente definite. Tuttavia, gli studi hanno suggerito che le perdite in uno o più dei geni RB1, RCBTB2, DLEU1 e ITM2B , ma in particolare il gene RP1 , possono essere coinvolte nel contribuire allo sviluppo e/o alla progressione di MPL.
Diagnosi
La diagnosi di MPL dipende dalla presentazione clinica del tumore, dalla somiglianza istopatologica con il liposarcoma mixoide e, soprattutto, dall'assenza dei geni di fusione FUS-DDIT3 sn EWSR1-DDIT3 nelle sue cellule neoplastiche.
Trattamento e prognosi
Mentre gli individui con MPL sono stati trattati con resezione chirurgica per rimuovere i loro tumori, una revisione del 2021 ha rilevato che non c'erano raccomandazioni di consenso per lo standard di cura per MPL rispetto ai regimi di radioterapia e chemioterapia (se usati da soli o in combinazione con la chirurgia) per curare questi tumori.
Istopatologia dei liposarcomi
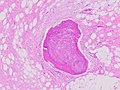
Fig. 1 Micrografia della formazione ossea in un tumore liposarcoma

Fig. 2 Micrografia di un tumore liposarcoma dedifferenziato

Fig. 3 Micrografia a bassa potenza del tumore del liposarcoma mixoide

Fig. 4 Micrografia ad alta potenza del tumore del liposarcoma mixoide


.jpg)
.JPG)
Imaging medico
L'ecografia medica e la risonanza magnetica (MRI) dei liposarcomi sono utili e spesso essenziali per determinarne l'estensione, l'accessibilità chirurgica e la relazione con eventuali disfunzioni d'organo osservate. Poiché l'ecografia di solito non è in grado di distinguere un liposarcoma da un lipoma benigno, la risonanza magnetica è l'imaging iniziale di scelta per prove comprovate relative a questa distinzione.

Fig. 5 Ecografia di un liposarcoma con aree ad alta eco riflesse dalla sua matrice lipomatosa e aree a bassa eco riflesse dalle sue aree non lipomatose.
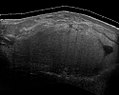
Fig. 6 Ecografia di un liposarcoma che mima un lipoma. Questa massa omogenea ad alta eco ha lo stesso aspetto di un lipoma.

Fig. 7 Risonanza magnetica di liposarcoma mixoide di alto grado, nella regione ascellare sinistra di un uomo di 40 anni, evidenziato dal suo colore bianco, in questa sezione orizzontale del tumore.



Società e cultura
Casi notevoli
- Chad Brown (1961-2014), un giocatore di poker, è morto di liposarcoma
- Richard Feynman (1918-1988), un fisico teorico, morì dopo un intervento chirurgico per affrontare la malattia.
- Rob Ford (1969-2016), ex sindaco di Toronto e consigliere comunale di Toronto, è morto di liposarcoma pleomorfo.
- Hokie Gajan (1959-2016), ex running back dei New Orleans Saints e commentatore radiofonico della squadra, è morto di liposarcoma.
- Charlie Davies (1986-), ex calciatore per la Philadelphia Union of Major League Soccer, diagnosticato un liposarcoma nel 2016.
- Mark Strand (1934-2014), ex poeta statunitense e vincitore del premio Pulitzer, è morto di liposarcoma.
Guarda anche
- lipoma
- The Wendy Walk , organizzazione no-profit la cui missione è raccogliere fondi e far conoscere i sarcomi, compreso il liposarcoma
Riferimenti
link esterno
| Classificazione | |
|---|---|
| Risorse esterne |